
| Program |
Platforms |
Source code
available |
Input formats |
Graphic
output formats |
Circular view |
Linear view |
Real time navigation |
Multiple genomes |
Annotation editing and creation |
Annotation searching |
Sequence searching |
| Genomorama |
OS X,
Windows, Linux |
√ |
EMBL,
GBK, ASN.1, FASTA, PTT |
PS, GIF |
√ |
√ |
√ |
√ | √ |
√ |
|
| Artemis |
Java |
√ |
EMBL,
GBK, FASTA, GFF |
JPG, PNG |
√ |
√ |
√ (via ACT) |
√ |
√ |
√ |
|
| Sockeye |
Java |
EMBL (via
server), GFF |
JPG |
√ |
√ |
√ |
|||||
| Apollo |
Java |
√ |
GAME XML,
GFF, GBK, EMBL, FASTA |
PS |
√ |
√ |
√ |
√ |
√ |
||
| Argo |
Java |
GFF, GBK,
GENSCAN, BLAST |
Printer |
√ |
√ |
√ (2) |
√ |
√ |
√ |
||
| IGB |
Java |
√ |
GFF,
FASTA, PSL, DAS |
Printer |
√ |
√ |
√ |
√ |
√ |
||
| GeneViTo |
Java |
PTT+FFN+FNA |
JPG |
√ |
√ |
√ |
√ |
√ |
|||
| CGView |
Java |
√ |
PTT, XML |
PNG, JPG,
SVG |
√ | ||||||
| GenoMap |
Tcl/Tk |
√ |
GRS |
PS |
√ |
||||||
| Genome2D |
Windows |
GBK,
FASTA, GLIMMER, PARADOX |
Printer,
WMF, BMP |
√ |
√ |
√ |
√ |
√ |
√ |
||
| GenomeComp |
Perl/Tk |
√ |
EMBL, GBK, FASTA | PS |
√ |
√ |
√ (2) |
√ |
|||
| GRS |
Linux,
Dec, Solaris |
√ |
GRS |
Printer |
√ |
√ |
√ |
√ |
|||
| Mauve |
Java |
√ |
GBK,
FASTA, SEQ |
PNG, JPG |
√ |
√ |
√ |
||||
| GATA |
Java |
√ |
GFF |
PNG |
√ |
√ |
√(2) |
||||
| GenomeViz |
Tcl/Tk/Perl
(no Windows) |
√ |
tab
delimited |
PS |
√ |
√ |
√ |
√ |
|||
| GenomePlot |
Tcl/Tk/Perl |
√ |
tab
delimited |
PS, GIF,
TIFF, JPG |
√ |
√ |
√ |
||||
| Bluejay |
Java |
XML |
Printer,
SVG |
√ |
√ |
√ |
|||||
| SeqVISTA |
Java |
√ |
EMBL,
FASTA |
JPG |
√ |
√ |
√ |
√ |
√ |
||
| DNAvis |
Windows,
Linux |
GFF, FASTA |
√ | √ | √ | ||||||
| AlignScope |
Java |
PTT + (BLAST
| GAME) |
√ | √ | √ (2) | ||||||
| Genome Workbench |
OS
X,
Windows, Linux |
√ | ASN.1, XML,
FASTA, GFF |
√ | √ | √ | √ | √ |
| Program |
Platforms |
Source code
available |
Graphic
output formats |
Circular view |
Linear view |
Real time navigation |
Multiple genomes |
Annotation editing and creation |
Annotation searching |
Sequence searching |
| Microbial Genome Viewer |
Browser-SVG |
√ |
Printer, SVG |
√ |
√ |
|||||
| cMap |
Java |
√ |
Printer |
√ |
√ |
√ |
√ |
|||
| ERGO Light |
Browser-HTML |
Printer |
√ |
√ |
||||||
| tair |
Brower-Java |
Printer |
√ |
√ |
||||||
| Jena Genome Viewer |
Browser-HTML |
PNG, PDF, PS |
√ |
√ |
√ |
√ |
||||
| Lightweight Genome Viewer |
Browser-HTML |
√ |
Printer |
√ |
||||||
| ChromoWheel |
Browser-SVG | Printer, SVG | √ |
|||||||
| GenDB |
Perl/Gtk |
√ |
PNG |
√ |
√ |
√ |
√ |
√ |
||
| BioVis |
Browser-SVG
(no Unix) |
√ |
Printer, SVG |
√ |
√ |
√ |
√ |
|||
| Vista |
Java |
√ |
PDF, JPG, GIF |
√ |
√ |
√ |
||||
| UCSC Genome Browser |
Browser-HTML |
√ |
PDF, PS |
√ |
√ |
√ |
| Vector formats (variable resolution) | Bitmap formats (fixed resolution) |
| SVG (Scalable Vector Graphics) | JPG (jpeg) |
| PS (Postscript) | PNG (Portable Network Graphics) |
| PDF (Portable Document Format) | GIF (Graphics Interchange Format) |
| WMF (Windows MetaFile) | BMP (Windows Bitmap) |
| Windows |
binarya |
sourceb |
| OS X |
binary (powerPC)a
binary(Intel)a |
sourceb (for
XCode
1.1) |
| Linux (Unix) |
sourceb |
| NCBI |
ASN.1 |
*.asn, *.val |
Both ASCII and binary formats
are supported |
| GenBank Flat File (US) |
GBK |
*.gbk |
Supported |
| GenBank Flat File (Europe/EMBL) |
EMBL |
*.embl |
Supported |
| Nucleic acid sequence |
FASTA |
*.ffn, *.fna |
Supported; Each sequence in a
file is
assumed to correspond to a gene |
| Protein Table File |
PTT |
*.ptt |
Supported |
| Amino acid sequence |
FASTA |
*.faa |
Supported (export only) |
| Genome Annotation Markup Elements |
GAME XML |
Not supported | |
| GENSCAN (gene predicition)
output file |
GENSCAN |
Not supported | |
| GLIMMER (gene prediction) output
file |
GLIMMER | Not supported | |
| Extensible Markup Language |
XML |
Not supported | |
| tab delimited (application
specific) |
tab delimited |
Not supported | |
| GRS input file |
GRS |
Not supported | |
| General Feature Format |
GFF |
Not supported | |
| Distributed Annotation System |
DAS |
Not supported | |
| BLAT output |
PSL |
Not supported | |
| Gene Transfer Format | GTF |
Not supported | |
| DNASTAR SEQ Format |
SEQ |
Not supported |



| Display
width (in bases) |
Displayed
objects |
Example |
Gene-centric Genome Display |
||
| width
> 15000 |
Genes/Pseudo
genes/RNA |
 |
Annotation size-centric Genome Display |
||
| width > 15000 | Annotations
greater than the length (i.e. size) of the median genome annotation |
 |
|
|
||
| 15000
> width > 70 |
All annotations | 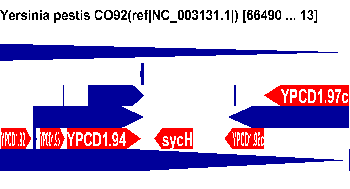 |
| width
< 70 |
Sequence |
 |
| Command |
Windows |
OS X |
Linux |
| Scroll left |
Left
arrow |
||
| Scroll right | Right
Arrow |
||
| Scroll up |
Page
Up |
||
| Scroll down |
Page
Down |
||
| Zoom in |
Up
Arrow |
||
| Zoom out |
Down
Arrow |
||
| Fit genome to window and center
at base 0 |
Ctrl+r |
Command+r |
Ctrl+r |
| Search dialog |
Ctrl+f |
Command+f |
Ctrl+f |
| Search again |
Ctrl+g, F3 |
Command+g |
Ctrl+g, F3 |
| Edit Annotation Dialog |
Ctrl+e |
||
| Select all genomes |
Ctrl+a |
Command+a |
Ctrl+a |
| Clear all selections |
Esc |
||
| Complement selected genome |
Ctrl+i |
Command+i |
Ctrl+i |
| Shift selected genomes up |
Shift+Up
Arrow |
||
| Shift selected genomes down | Shift+Down
Arrow |
||
| Increase the number of genomes
per page |
+ |
||
| Decrease the number of genomes
per page |
- |
||
| Jump to base number 'N' (just
like the vi text editor) |
'N'
Shift+g |
||
| Magnify the annotation currently
under the pointer |
Ctrl+m |
Command+m |
Ctrl+m |
| Quit the program |
Ctrl+q |
Command+q |
Ctrl+q |
| Command |
Windows and
Linux |
OS Xa |
| Scroll Left |
Left Button
click and drag |
[Left] Button click and drag |
| Scroll Right |
Left Button click and drag | [Left] Button click and drag |
| Scroll All Genomes (Left/Right/Up/Down) |
Middle
button click and drag |
Middle button click and drag |
| Zoom In |
Right button
click and drag, Wheel |
Right button click and drag, Wheel |
| Zoom out |
Right button click and drag, Wheel | Right button click and drag, Wheel |
| Select genome |
Left button
click |
[Left] Button click |
| Toggle select multiple genomes |
Ctrl+Left button click | Command + [Left] Button click |
| Genome information |
Left button double click | [Left] Button double click |
| Annotation information |
Left button double click | [Left] Button double click |
| Display sequence: Set first base |
Shift + Left button click | Shift + [Left] Button click |
| Display sequence: Set last base |
Shift + Left button click | Shift + [Left] Button click |

| %G+C = |
(Gf + Cf)/(Af
+Tf + Gf + Cf + Nf) |
| %A+T = |
(Af + Tf)/(Af +Tf + Gf + Cf + Nf) |
| GC Skew = |
(Gf - Cf)/(Gf + Cf) |
| AT Skew = |
(Af - Tf)/(Af + Tf) |
| Keto Skew = |
(Gf + Tf - Af - Cf)/(Af +Tf + Gf + Cf + Nf) |
| Purine Skew = |
(Gf - Tf + Af - Cf)/(Af +Tf + Gf + Cf + Nf) |
| Query Target |
Query String |
Notes |
| Annotation
Keyword(s) |
one or
more text strings |
Keyword
searches are case insensitive and do not depend on the order of query
strings. |
| gi |
number |
Compare
input number against the gi of all loaded annotations. |
| Locus
Name/Tag |
string |
Compare input string against both locus name and tag of all loaded annotations. |
| DNA
sequence string |
string
(characters can be A, T, G or C) |
|
| Probe
hybridization |
string (characters can be A, T, G or C) | |
| PCR
hybridization/extension |
pair of strings (characters can be A, T, G or C) |

| Selected annotations |
Yellow |
| Background |
White |
| Text |
Black |
| Gene |
Red |
| Pseudo-gene |
Pink |
| CDS |
Green |
| Misc annotations |
Blue |
| RNA |
Black |
| Intergenic space |
Grey |
 |
Operated by the Los Alamos National Security, LLC for the National Nuclear Security Administration, of the US Department of Energy. Copyright © 2005 LANSLLC | Disclaimer/Privacy |